Kassidi Petersen, Molly Ross, Molly Felix, Lauren Grillo, Cassandra Marshall, Dr. Julia Oxford

Introduction
Background information about the disease Sanfilippo syndrome (also known as MPSIII) type A is caused by the mutation of the gene encoding SGSH, N-sulfoglucosamine sulfohydrolase, on chromosome 17, region 17q25.3
It is a recessive gene that affects around 1 in 70,000 babies each year
This mutation affects the mucopolysaccharides and causes the patient to lose an enzyme that is crucial to breaking down heparan sulfate
Heparan sulfate is stored in the cells instead of breaking down and causes progressive damage
Symptoms become apparent after the age of 1-years-old
- Learning abilities decline after the ages of 2 and 6
- The symptoms and progression rate are different in each patient
- Limit of physical growth, resulting in small stature
Other common symptoms include:
- Behavioral problems
- Coarse facial features
- Full lips
- Heavy brows
- Sleep difficulties
- Stiff joints
- Difficulty walking
Diagnosis includes a urine test looking for high levels of mucopolysaccharides

Prospective Treatment
Enzyme Replacement Therapy and Outcomes
Enzyme replacement therapy has gone through multiple trials to see if testing different ways to replace the missing enzyme that inhibits the degradation of MPS.
- Animal models of enzyme therapy
- Protein fusion and direct administration to CNS have been proven effective
- Problems are a high concentration of enzymes are needed and fusion to CNS is highly invasive and may not work on children, the main affected population
- Clinical enzyme replacement trials
- Began in 2016 with 12 patients
- In the trial recombinant human heparan-N-Sulfatase has been administered intrathecally to each of the patients monthly
- 4 showed a decline in development 6 remained stable and 2 were inconclusive
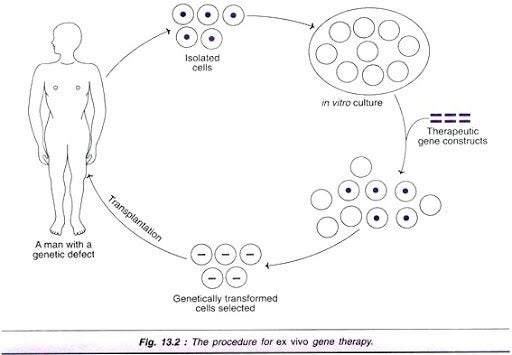
Gene therapy in Sanfilippo Syndrome Type A is used in hopes that the stem cells that are introduced from the bone marrow of a healthy individual will be catalytically active and this activity will spread to native cells so that they will be catalytically active too.
- Preliminary testing in mice showed significant decrease in activity after treatment if treated early.
Mice treated with a Sulfate Modifying Factor 1 in the SGSH gene using an internal ribosomal entry site (SGSH-IRES-SUMF1) showed activity similar to normal mice. Early treatment by injection was sufficient in prevention or delay of central nervous system issues in mice with Sanfilippo Syndrome. This study and others like it allowed human clinical trials to begin. (Fraldi A, et al.)
- In human trials, a similar result was found. The best outcomes are from younger patients who received treatment early. During the trials, patients showed no negative side effects at the injection site. ( Tardieu M,et al.)
Treatment for this disease using stem cells in bone marrow is still in trials but looks hopeful.
Conclusion
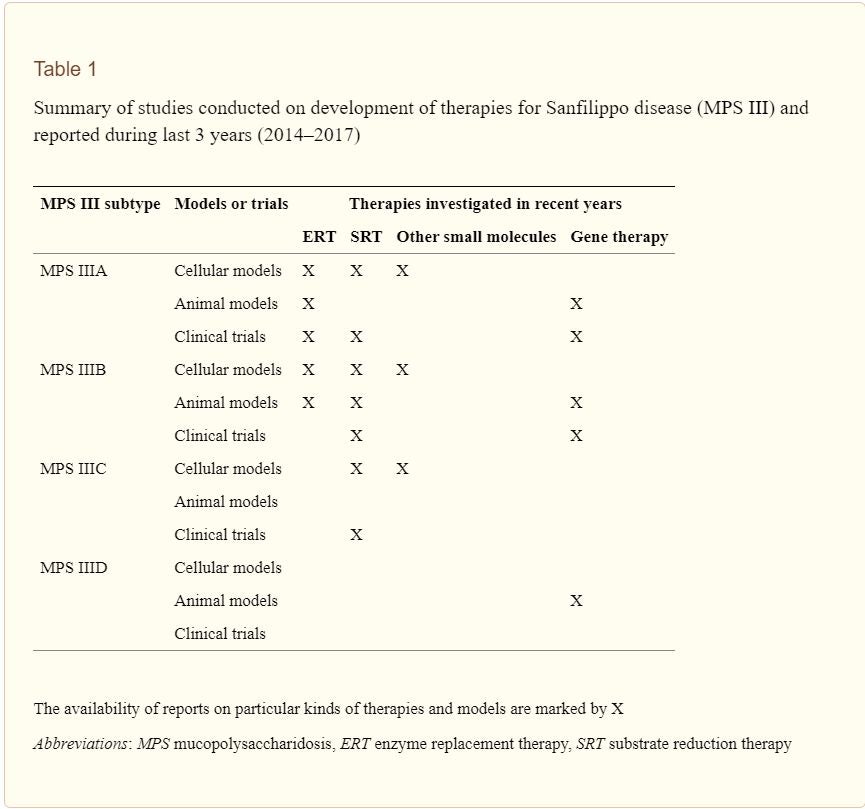
Overall, treatments for Sanfilippo syndrome type A are still in the preliminary stages. Enzyme replacement therapy and gene therapy currently are at the forefront of research. The advancement of clinical trials to human subjects will be of principal interest as research continues. MPS III is a syndrome associated with high lethality: it is crucial that therapies are effective for young individuals. Due to this, our meta-analysis found gene therapy to be a more promising avenue of treatment. Future research should heavily invest in gene therapy to improve outcomes for affected individuals.
Psychological and Cultural Considerations
Psychological Symptom Management:
Sanfilippo syndrome presents initially with psychosomatic symptoms, similar to those seen in individuals with ADHD. Research into management of these symptoms is critical to patient wellbeing as well as elevating quality of life.
Cultural Considerations:
Attention to areas of prevalence should also be taken into account with this syndrome. Geographic distribution of Sanfilippo shows a high density of cases taking place in Latin America.
Citations
- Fraldi A, Hemsley K, Crawley A, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16:2693–2702.
- Tardieu M, Zérah M, Husson B, et al. Intracerebral administration of adeno-associated viral vector serotype rh. 10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther. 2014;25:506–516
- “MPS III.” MPS Society, mpssociety.org/learn/diseases/mps-iii/.
- Gaffke, Lidia et al. “How close are we to therapies for Sanfilippo disease?.” Metabolic brain disease vol. 33,1 (2018): 1-10. doi:10.1007/s11011-017-0111-4 “Sanfilippo Children’s Foundation.” Sanfilippo, www.sanfilippo.org.au/research/therapy-avenues/enzyme-replacement-therapy-clinical-trials.
Additional Information
For questions or comments about this research, contact Kassidi Petersen at kassidipetersen@u.boisestate.edu.