Chloe Watt, Leyla Cufurovic, Joe Peterson, Janae Stockton, Dr. Julia Oxford

Abstract
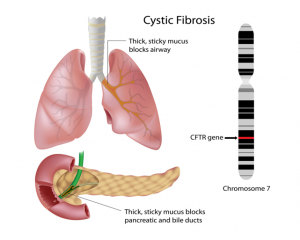
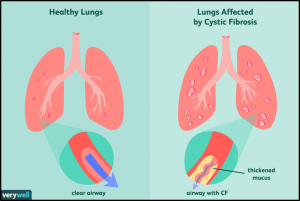
Cystic fibrosis was first recognized as a new disease in 1938 when autopsies of children who died from malnourishment revealed similar findings. Currently, there are approximately 30,000 people living in the U.S. who are affected by this rare genetic disease. Cystic fibrosis is a congenital progressive disorder caused by a mutation in the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). Mutations in the CFTR gene cause the mucus in the body to become thick and glue-like, blocking ducts and tubes distributed within the body. Buildup in mucus can cause a multitude of problems, eventually leading to death. Treatment of an individual with cystic fibrosis depends on the severity of symptoms. Antibiotics can prevent and treat chest infections. Other medications can also treat thick mucus in the lungs by thinning and hydrating the mucus, allowing it to clear and also widening the airways to make breathing easier. Lung transplant may be needed in the case of lung damage. The FDA recently approved Trikafta, the first triple combination therapy available to treat CF. Here, we explore the manner in which this trifecta of drugs helps the protein encoded by the CFTR gene mutation function more effectively.
Diagnosis
- Genetic testing to detect mutated CFTR genes. This test can confirm a positive cystic fibrosis screening test and sweat test. If genetic testing is done as part of newborn or other screening, it may not be repeated during the newborn stage.
- Prenatal diagnostic tests to diagnose cystic fibrosis in an unborn baby, using mutated CFTR genes. This is done with procedures that take either a sample of amniotic fluid, the liquid in the sac surrounding the unborn baby, or tissue from the placenta. Cells from these samples are checked for gene mutations.
- Sweat test for high sweat chloride to see if a patient has high levels of chloride in the sweat. The sweat test is the standard test for diagnosing cystic fibrosis. It may be used if someone has symptoms that may indicate cystic fibrosis, or to confirm a positive diagnosis from a screening of a newborn baby.
- Measuring nasal lining involves running a small electrical current across the epithelium. Different solutions are applied to the nasal lining and the electrical current is measured. People with CF respond very differently than those without CF to this test.
Clinical Description
- Cystic fibrosis (CF) is an inherited disorder that causes severe damage to the lungs, digestive system and other organs in the body.
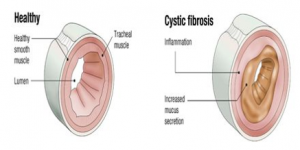
- Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in people with CF, a defective gene causes the secretions to become sticky and thick.
- Symptoms in newborns may include delayed growth, failure to gain weight normally during childhood, no bowel movements in the first 24-48 hours of life, and salty skin.
- Symptoms that may be noticed later in life are infertility in men, repeated inflammation of the pancreas i.e. pancreatitis, respiratory symptoms, and clubbed fingers.
- Cystic fibrosis is caused by mutations in a gene called the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The CFTR gene provides instructions for the CFTR protein. The CFTR protein is located in every organ of the body that makes mucus, including the lungs, liver, pancreas, and intestines, as well as sweat glands.
Prognosis
- Most children with CF stay in good health until they reach adulthood. They are able to take part in most activities and attend school. Many young adults with CF finish college or find jobs.
- Sadly, lung disease eventually worsens to the point where the person is disabled. Today, the average lifespan for people with CF who live to adulthood is about 37 years. Death is most often caused by lung complications.
- There currently is not a cure for CF and it cannot be prevented. However, new treatment methods help children who have CF have a better quality of life. Advances in the treatment of CF have also increased the expected age of a patient’s survival.
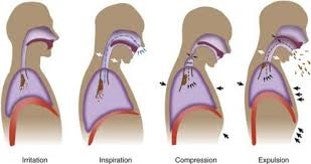
Unresolved Questions
- What are the current possibilities in a new gene therapy approach?
- Will we be able to repair and splice mutations in the CFTR gene through genome editing on a widespread scale?
- Is the new drug Trikafta the answer to helping patients, especially those of a young age, to dealing with and managing Cystic Fibrosis? According to researchers this new drug is a “game changer” and the opportunities this treatment provides to those beginning to grow up with CF is insurmountable.
Treatment Options
Starting at the age of five or older, a CF care team will have the patient take a breathing test known as pulmonary function test (PFT). This test measures breathing, coughing, and the amount of sputum being produced.
Airway clearance techniques (ACTs) loosen thick, sticky mucus so it can be cleared from the lungs by coughing or huffing. Some use percussion (clapping) based techniques to loosen mucus, while others use chest physical therapy (CPT) which uses gravity to drain mucus from the five lobes of the lungs.
- Inhaled bronchodilators are taken before ACTs to widen airways. Mucus thinners are taken during ACTs to move mucus out of airways. Inhaled antibiotics are taken after finishing the ACTs which allows medication to work more effectively.
Antibiotics are used to destroy of slow down growth of bacteria that cause infections in people suffering from CF. These can be taken orally, intravenously, or intramuscularly.
Lung transplantation can extend and improve their quality of life, but it involves an extensive evaluation process and a commitment to living the lifestyle required to keep their new lungs healthy.
Pancreatic enzyme replacement therapies are used to help the body absorb food and necessary nutrients.
Cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapies are designed to correct the malfunctioning protein made by the CFTR gene. This includes helping the CFTR protein form the right shape so that it can traffic to the cell surface. These therapies are also used to bind to the defective protein at the cell surface and open the chloride channel so that chloride can flow through, regulating the amount of fluids at the surface of the cell.
- Kalydeco, Orkambi and Symbdeko are current CFTRs, but are only effective in people with specific mutations.
- Trikafta is the newest CFTR modulator and has proven to be very effective as it is a triple combination of the other modulators.
- More potential CFTR modulators are in development to address the underlying cause of the disease in people with other CF mutations.
References
- “About Cystic Fibrosis.” CF Foundation. 2018.
- American College of Chest Physicians. “Diagnosing and Treating Cystic Fibrosis.” American Lung Association, 4 Apr. 2018.
- “Cystic Fibrosis.” National Heart Lung and Blood Institute, U.S. Department of Health and Human Services.
- “Cystic Fibrosis: MedlinePlus Medical Encyclopedia.” MedlinePlus, U.S. National Library of Medicine.
- Mayo Clinic Staff. “Cystic Fibrosis.” Mayo Clinic, Mayo Foundation for Medical Education and Research, 4 Feb. 2020.
- Pena, Ana. “Trikafta Will Be ‘Life-Transforming,’ Especially for Younger CF…” Cystic Fibrosis News Today, 14 Jan. 2020.
- “Treatments and Therapies.” CF Foundation.
- “Cystic Fibrosis Market 2018; Superior Growth at a CAGR of 13.5% | Cystic Fibrsosis Treatment are pushing the market growth globally by 2022 – Avail at MRFR.” Medgadget. 18 July 2018.
Additional Information
For questions or comments about this research, contact Chloe Watt at chloewatt@u.boisestate.edu.