Nick Cordell, Jordan Council, Rachel Kessinger, Crislyn Rausch, Stephen Schott, Dr. Julia Oxford

Abstract

Image credit: https://ghr.nlm.nih.gov/condition/mowat-wilson-syndrome
Mowat-Wilson syndrome (MWS) is a rare genetic disorder that causes systemic deficiencies and abnormalities during development. Common presentations of this disorder include Hirschsprung disease (HSCR), intellectual disability, delayed development, distinctive facial features, microcephaly, epilepsy, and heart defects. MWS is likely underdiagnosed, especially in patients who do not display HSCR. Prognosis for MWS depends on the severity and presence of congenital anomalies, and few patients have been reported to live into early adulthood. Therapy may also be required to control seizures, while occupational and speech therapy may help with delayed psychomotor development which is prevalent in all patients. MWS is caused by a pathogenic variant of the ZEB2 gene that follows an autosomal dominant pattern of inheritance. A mutation of the ZEB2 gene suggests that the myelination process may represent a potential therapeutic target. Current treatments focus on symptoms; however, a potential therapeutic target may focus on the effects the mutation has on gene expression.
Definition
Intellectual disability characterized by distinct facial structures and may present with or without Hirschsprung’s disease. MWS was discovered in 1998 by Dr. Mowat and Dr. Wilson. Prevalence of MWS is estimated between 1/50,000 to 1/70,000 live births, with over 300 cases reported so far.
Clinical Description
Common Clinical Features
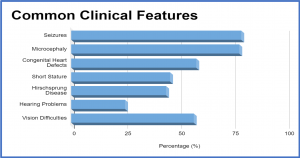
- Seizures – more than 75%
- Microcephaly – more than 75%
- Congenital Heart Defects – more than 50%
- Short Stature – less than 50%
- Hirschsprung Disease – less than 50%
- Hearing Problems – 25%
- Vision Difficulties – more than 50%
Facial Features
Narrow triangular chin, deep set large eyes, broad nasal bridge, saddle nose, rounded nasal tip, open mouth, full or everted lower lip, posteriorly rotated ears, and large uplifted ear lobes with a central depression.
Nervous System
Seizures, intellectual disability, dysphagia
Musculoskeletal System
Microcephaly, short stature, slender body type, calcaneovalgusdeformity.
Cardiovascular System
Congenital heart defects: abnormalities in pulmonary arteries and/or valves
Genitourinary System
Renal abnormalities, in male’s hypospadias and cryptorchidism.
Digestive System
Hirschsprung disease (HSCR) and constipation
Brain Structure Deformities
Cerebral atrophy, underdeveloped hippocampus, and deformities of the temporal and frontal lobes.
Etiology
MWS is a genetic mutation affecting the ZEB2 gene located on chromosome 2. Mutations range from missense to deletions or insertions. MWS follows an autosomal dominant pattern of inheritance, one abnormal gene will cause this syndrome. Occurring de novo, neither parent is seen to have any mutation.
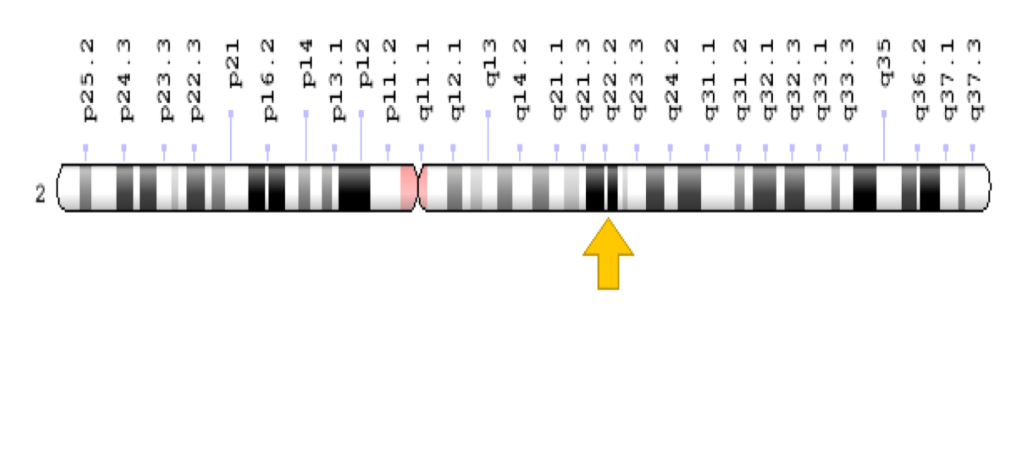
Image Credit: Genome Decoration Page/NCBI
Diagnosis
- Diagnosed during infancy or childhood
- Identification of characteristic physical findings and facial appearance
- Computerized tomography (CT) scanning or magnetic resonance imaging (MRI) of the brain, kidney ultrasound or heart ultrasound
- Molecular genetic testing for mutations in the ZEB2 gene.
- Prenatal diagnosis for parents with an affected child
Differential Diagnosis
- Diagnosed during infancy or childhood
- Identification of characteristic physical findings and facial appearance
- Computerized tomography (CT) scanning or magnetic resonance imaging (MRI) of the brain, kidney ultrasound or heart ultrasound
- Molecular genetic testing for mutations in the ZEB2 gene.
- Prenatal diagnosis for parents with an affected child
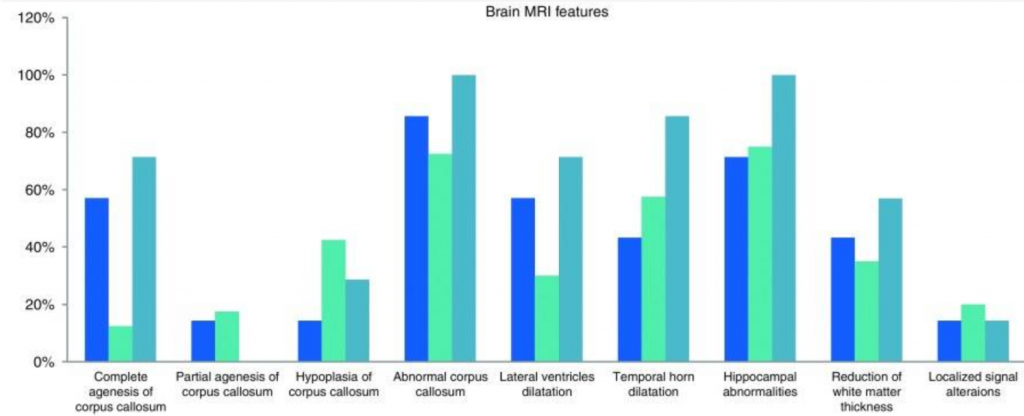
Image Credit: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5438871/
Prognosis/Management
- Few patients reported to live into adulthood
- Prognosis depending on severity of congenital anomalies
- No cure, only treatments for symptoms
- Surgery/ physical therapy for somatic symptoms (including heart, intestinal, and vascular defects)
- Speech therapy and Supportive Care
- Pharmaceuticals for constipation, GERD, and seizures
Unsolved Questions
- Other genes are involved in the agenesis of corpus callosum?
- Cause of de novo mutations?
- How are symptoms connected to deletion/mutations of ZEB2?
- Comprehensive picture of ZEB2 regulatory elements.
References
- Adam MP, ContaJ, Bean LJH. Mowat-Wilson Syndrome. 2007 Mar 28 [Updated 2019 Jul 25]. In: Adam MP, ArdingerHH, PagonRA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1412/
- Mowat, D.R., Wilson, M.J., & Goossens, M. (2003). Mowat-Wilson syndrome.Journal of Medical Genetics, 40(5), 305-310. doi: 10.1136/jmg.40.5.305
- Garavelli, L., Mainardi, P.C. Mowat-Wilson syndrome. OrphanetJ Rare Dis 2, 42 (2007). https://doi.org/10.1186/1750-1172-2-42
Additional Information
For questions or comments about this research, contact Nicholas Cordell at nicholascordell@u.boisestate.edu.