Darren Lighter, Grace Coughlin, Isreal Anaya Carmona, Karen Sanchez, Dr. Julia Oxford

Background
Baller-geroldsyndrome (BGS) triggers the malformation of the cranium along with the disfigurement or complete absence of the distal zones of the upper limbs, in which the only operable treatment is risky surgery of the skull that may permanently devastate mental functions. Furthermore, BGS deteriorates the cardiovascular, respiratory, and immune system culminating in an atrocious complex of burdensome symptoms. Further research could help prevent BGS or improve the life of those affected by this syndrome.
Epidemiology
Worldwide, less than 1 per 100,000 people are diagnosed with BGS. Less than 40 cases have been disclosed in medical literature.
(Van Maldergem, L, et. al. 2018)
Clinical Description
Major Abnormalities
- Craniosynostosis: the joints between the baby’s skull fuse prematurely resulting in a misshapen skull
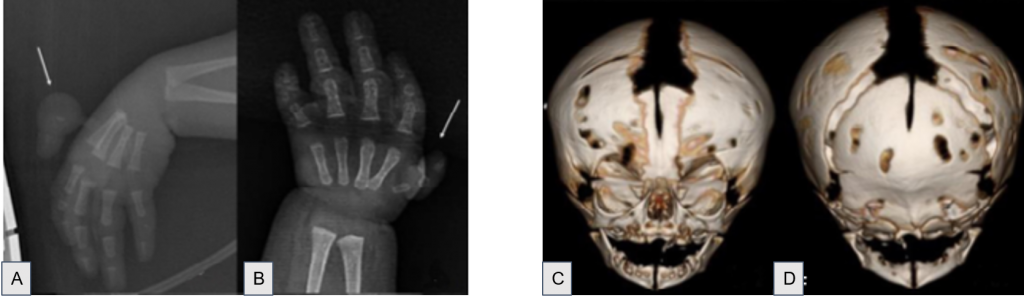
- Limb abnormalities: radial aplasia and absent or malformed thumbs
- Cardiac Defects: ventricular or atrial septal defects
Minor Abnormalities
Anal and urogenital defects, central nervous system defects, and mental retardation (Savarirayan 1998)
Aetiology
- Genetic based growth deficiency syndrome
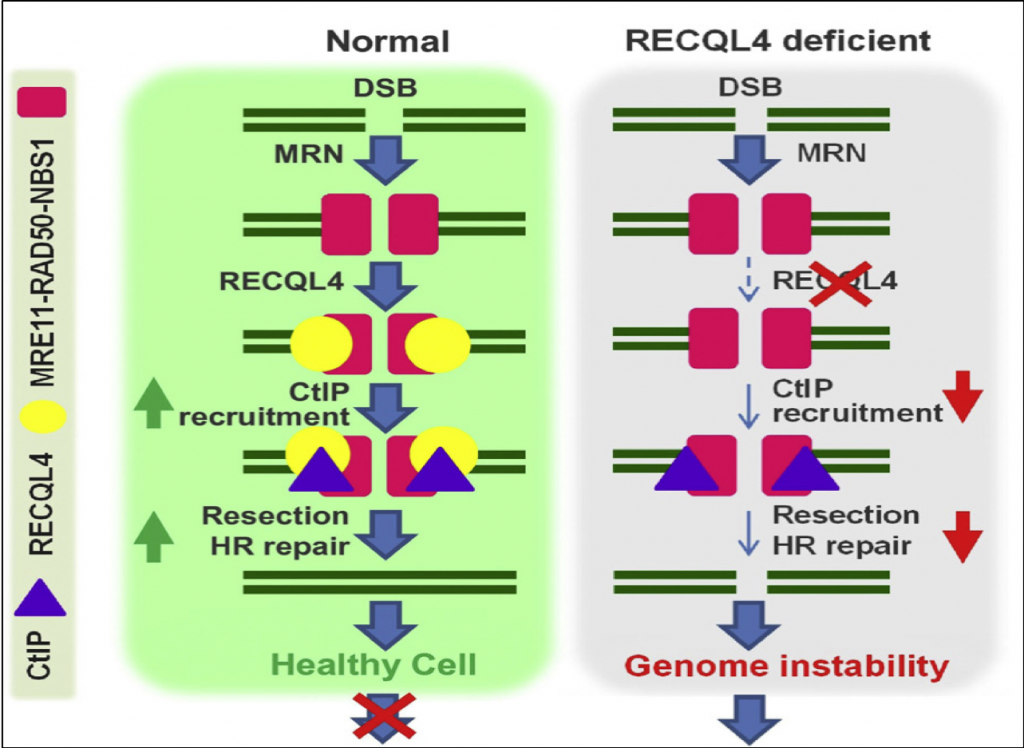
- Caused by a mutation in the RECQL4 gene.
- RECQL4 gene encodes for the RECQL4 protein in which shortage of the protein is known to prevent DNA replication and repair.
- Gene mutation inherited in an autosomal recessive pattern (NIH 2020)
RECGL 4 Gene Mutation
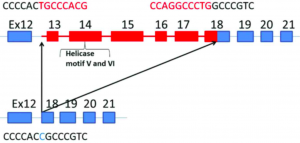
Figure 2. Model of genetic deletion and confirmation of deletion in patient through exon sequencing and PCR. (Hideo Kaneko, et. al 2017)
Common Baller-Gerold Mutation
A) Model showing the deletion of exons 13-17 leading to an inactivated helicase protein. B) Model showing the cascading effects of the inactivated RECQL4 helicase in double-stranded break repair. C) PCR confirming exon deletions in patient. (Kaneko .H, et. al. 2017)

Differential Diagnosis
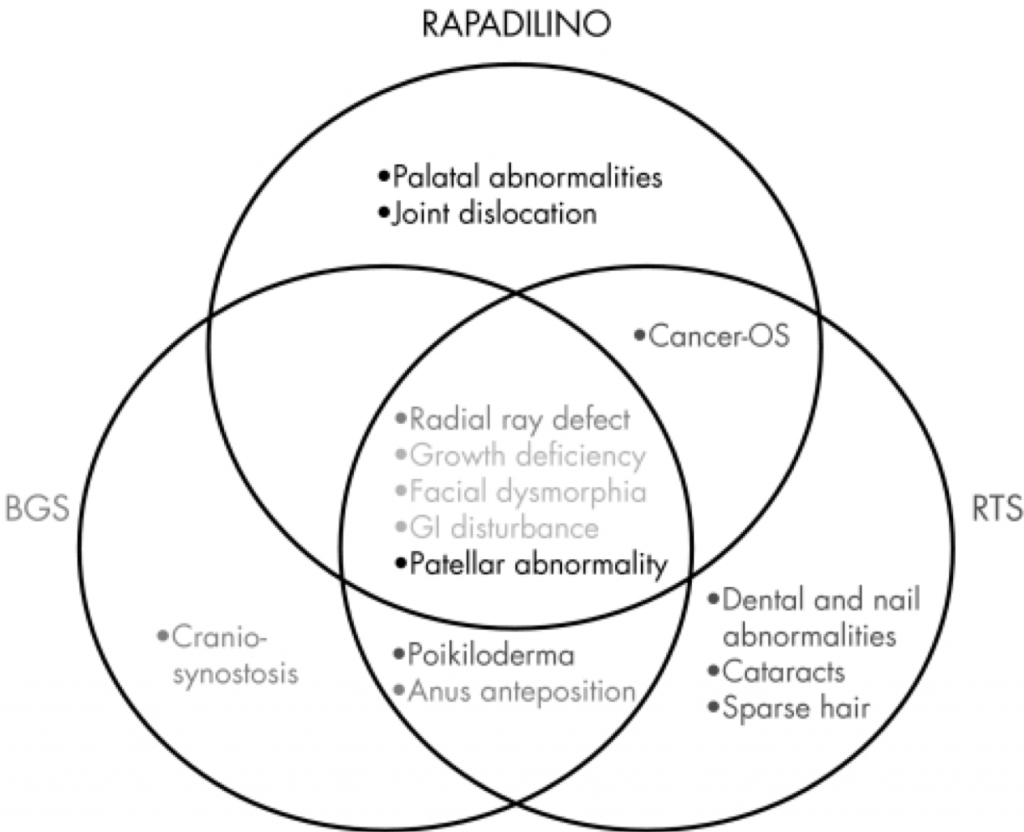
BGS Only
- Cranio-synostosis
BGS and RTS
- Poikiloderma
- Anas anteposition
RTS Only
- Dental and nail abnormalities
- Cataracts
- Sparse hair
RTS and Rapadilino
- Cancer-OS
Rapadilino Only
- Palatal abnormalities
- Joint dislocation
All three diseases
- Radial ray deflect
- Growth deficiency
- Facial dysmorphia
- GI disturbance
- Patellar abnormaility
Antenatal Diagnosis
- Serial ultrasound can help identify abnormalities such as limb shortening, radial hypoplasia/aplasia, and brachycephaly. These abnormalities can be detected at 14 weeks of gestation.
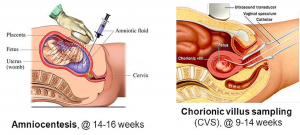
- Amniocentesis: DNA extracted from the fetal cells which can be done in pregnancies with a high risk for BGS. It is usually performed between 14-16 weeks of gestation.
- Chorionic villus sampling: small piece of placental tissues is analyzed. It is performed between 9-14 weeks of gestation and can be done transcervical or transabdominal. (Faivre 2009)
Management/Treatment
(Wang, L.L, et. al. 2003)
Management: Consultations following the initial diagnosis
- Dermatology to evaluate for poikiloderma
- Clinical geneticist or genetic counselor for comprehensive genomic testing and gene targeted testing.
- Orthopedic surgery to evaluate extremity functions for possible surgery
- Neurosurgery or a craniofacial specialists to evaluate craniosynostosis
- Oncologists to evaluate for cancer development
- Preventative measures for further complications such as osteosarcoma and lymphoma
- Limitation of UV radiation to protect for possible skin cancer if poikiloderma is present
Treatment: Currently, there is no cure. There are only symptomatic treatment options available.
Surgery to treat craniosynostosis is usually performed before 6 months of age. Hand surgery to create a thumb from an existing digit, (pollicization of the index finger) is performed to help with grasping function. (Maldergem et al. 2007)
Discussion
- Based on case study information, the general age of patients that undergo treatment are between ages 2 and 4. An unresolved question is this disorder generally leads to death at an early age or if treated early enough, can patients live to adulthood.
- Future research can be performed to define additional signs and symptoms that distinguish BGS from similar syndromes in addition to the broad phenotype of craniosynostosis and which involve the RECQL4 gene. Further studies on effective genetic treatments such as CRISPR may be beneficial as well.
- See citation links for References
Additional Information
For questions or comments about this research, contact Grace Coughlin at gracecoughlin@u.boisestate.edu.